Python Example Connections.py
About This Example:
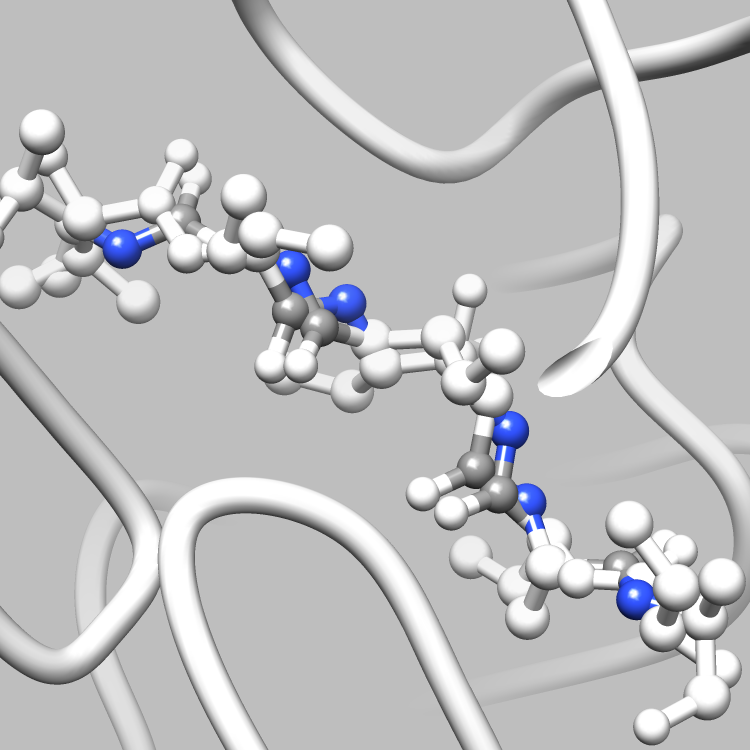
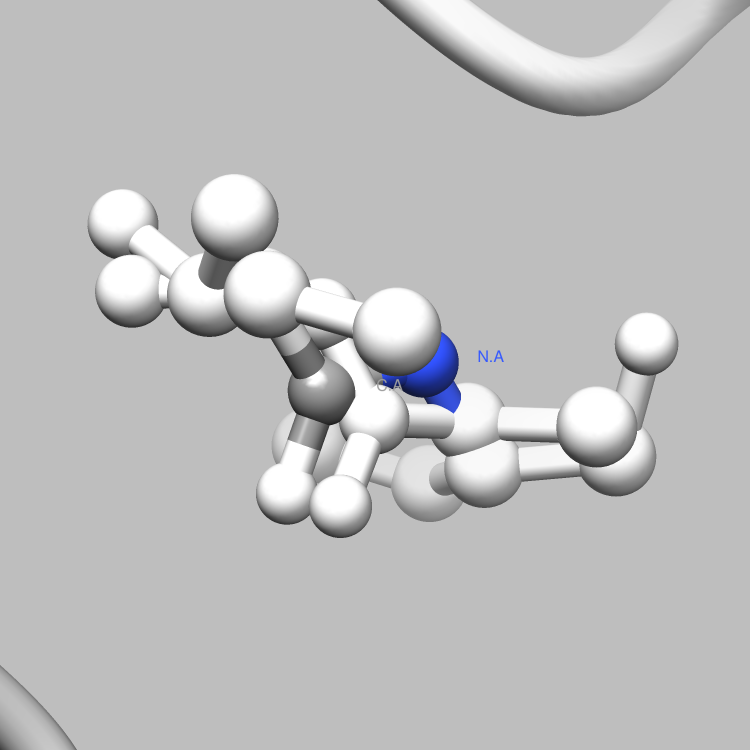
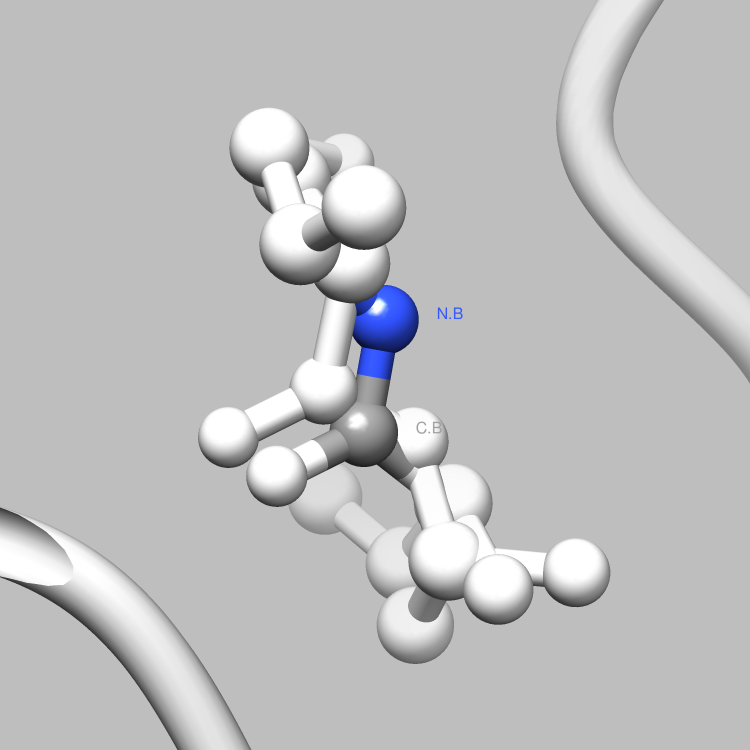
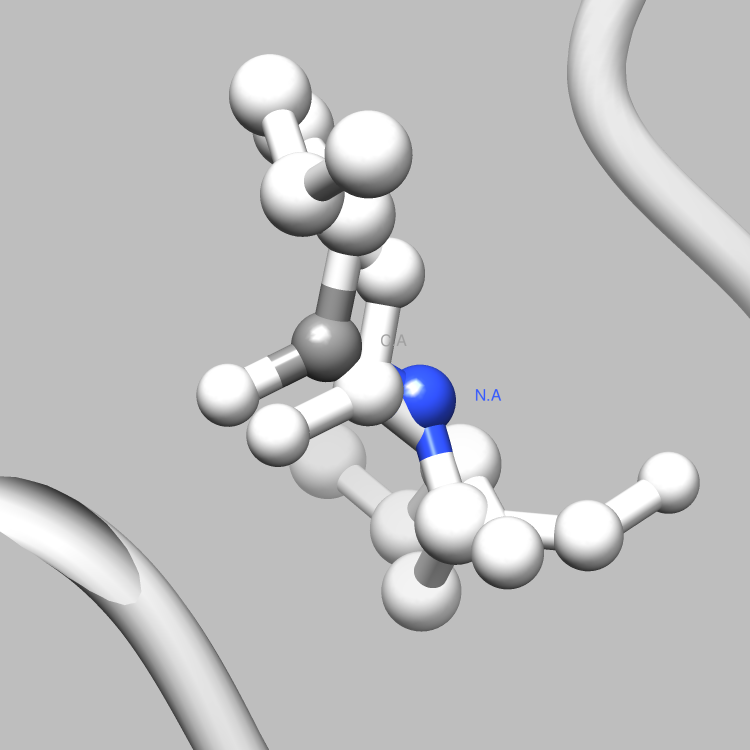
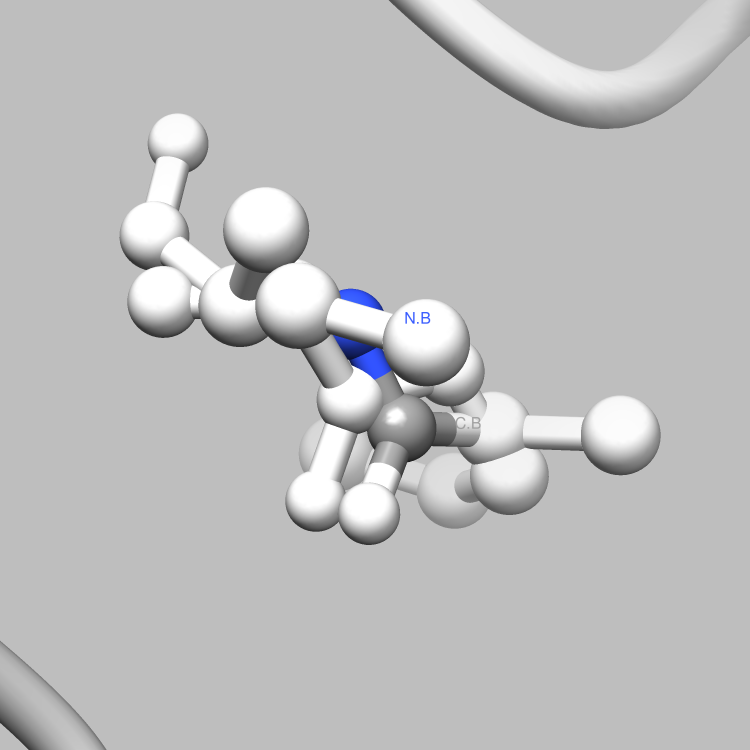
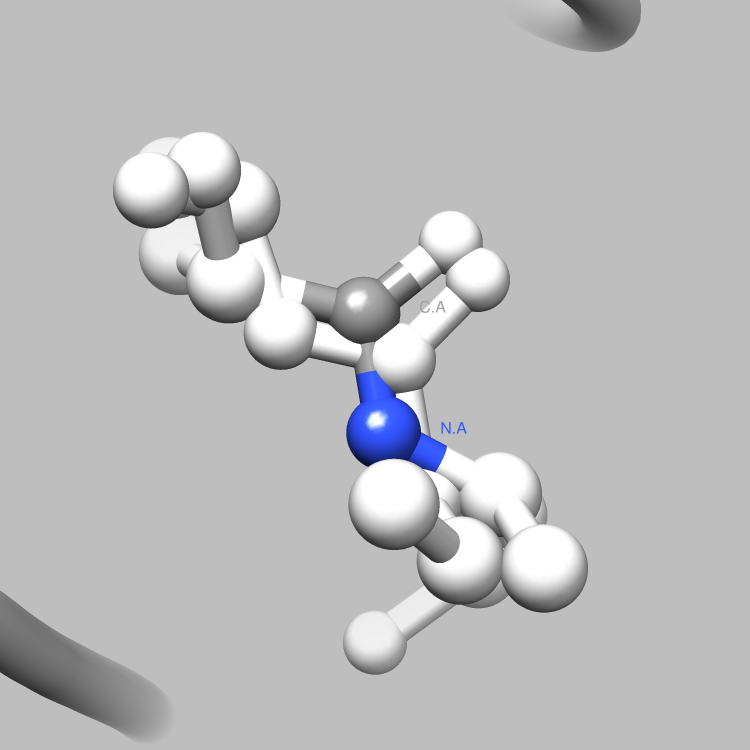
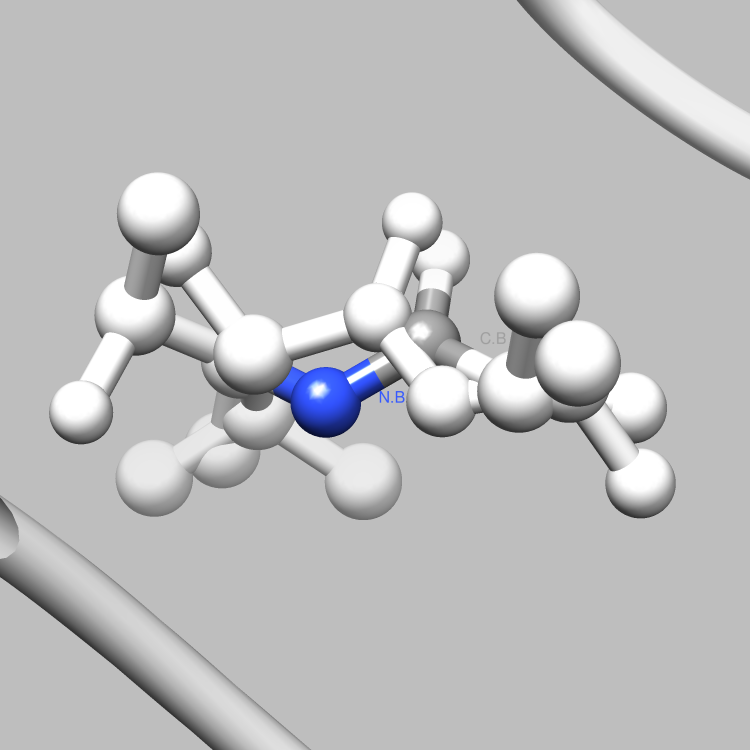
This example is one of a few that shows how the PDBX library can be used to interface with Chimera, such that useful and interesting aspects of a molecule, e.g., certain connections among certain atoms, obtainable via parsing CIF files, can be located and used as the subject of a Chimera render or animation. This particular example shows how to retrieve and iterate over the struct_conn category, which delineates linkages in a molecule, and locate these linkages of interest (in this case, covalent bonds) for Chimera to display and animate. It is easy to extend this example, say, to handle a set of connection types of interest, each to be displayed in a different color in Chimera, or to focus on connection types of interest only among certain atoms of interest.
Build Instructions:
Files: Connections.py, 5HVP.cif, Connections.sh
Save Connections.py and the CIF data file
Run python Connections.py /path/to/file.cif, which generates a /path/to/5HVP.com file which you can open
with chimera /path/to/5HVP.com Alternatively, you can save the script with Connections.py, set the Chimera path, and
run ./Connections.sh /path/to/5HVP.cif, which will automate the process.
Methods To Note
from pdbx.reader.PdbxContainers import ContainerBase from pdbx.reader.PdbxContainers import DataCategory
getObj(self, name)Returns the DataCategory object specified by name.getRowCount(self)Returns the number of rows in the category table.getValue(self, attributeName=None, rowIndex=None)Returns the value of the attribute attributeName at row index rowIndex.
Example Source Code
"""
Connections.py
For some CIF file, generate a Chimera command (COM) file
to iterate through and animate each interatomic covalent
connection in the molecule.
Lines with superscriptions contain footnoted references or explanations.
"""
from os.path import splitext
from pdbx.reader.PdbxReader import PdbxReader
from pdbx.reader.PdbxContainers import *
from sys import argv, exit
def prepareOutFile(file, name) :
file.write("windowsize 500 500\n") # Set the window size to 500 x 500 px
file.write("open %s\n" % name) # Open the CIF file
file.write("preset apply pub 4\n") # Apply publication preset #4
file.write("color white\n") # Color the entire molecule white
file.write("set bg_color gray\n") # Color the background gray
file.write("repr bs\n") # Represent the atoms in ball-and-stick format
file.write("savepos fullview\n") # Remember this position (the full view of the molecule)
def writeConnection(selection, file) :
sel = " | ".join(selection)
file.write("sel %s\n" % sel) # Select the two partner atoms
file.write("color byelement sel; label sel\n") # Color them by element
file.write("sel sel za<3.0; wait 20\n") # Further select all atoms within 3.0 angstroms of the partner atoms
file.write("focus sel; wait 25; ~disp ~sel; wait 68\n"); # Focus in on the selection and hide all non-selected atoms
file.write("disp ~sel; ~label sel; reset fullview 20\n") # Return to the full molecule view
file.write("color white sel; ~sel; wait 20\n") # Uncolor and drop the selection
# Check for improper usage
if len(argv) != 2 :
exit("Usage: python Connections.py /path/to/file.cif")
# Open the CIF file
cif = open(argv[1])
# A list to be propagated with data blocks
data = []
# Create a PdbxReader object
pRd = PdbxReader(cif)
# Read the CIF file, propagating the data list
pRd.read(data)
# Close the CIF file, as it is no longer needed
cif.close()
# Retrieve the first data block
block = data[0]
# Retrieve the struct_conn category table, which delineates connections1
struct_conn = block.getObj("struct_conn")
# Use the CIF file pathname to generate the Chimera command file (.COM) pathname
(file, ext) = splitext(argv[1])
comFileName = file + ".com"
# Open the COM command file for writing
comFile = open(comFileName, 'w');
# Write out some basic Chimera initialization commands
prepareOutFile(comFile, argv[1])
# Iterate over the rows in struct_conn, where each row delineates an interatomic connection
for index in range(struct_conn.getRowCount()) :
# A container to hold each partner atom's Chimera selection string
selection = []
# Verify that the connection is covalent2
if struct_conn.getValue("conn_type_id", index) == "covale" :
# Analyze the current row twice, once per partner
for partner in ["ptnr1_", "ptnr2_"] :
# Add this atom's Chimera selection string to the container3,4
selection.append(":%s,%s.%s@%s.%s" % (struct_conn.getValue(partner + "auth_seq_id", index),
struct_conn.getValue(partner + "auth_comp_id", index),
struct_conn.getValue(partner + "auth_asym_id", index),
struct_conn.getValue(partner + "label_atom_id", index),
struct_conn.getValue("pdbx_" + partner + "label_alt_id", index)))
# Write out commands for Chimera to customize the display of these connected atoms
writeConnection(selection, comFile)
# Write out the Chimera close command, as all connections have been processed
comFile.write("stop\n")
# Close the COM file
comFile.close()