Philip E. Bourne, Helen M. Berman, Brian McMahon, Keith D.Watenpaugh, John Westbrook and Paula M.D.Fitzgerald
Methods in Enzymology. 1997 277, 571-590.
The Protein Data Bank (PDB) format provides a standard representation for macromolecular structure data derived from X-ray diffraction and NMR studies. This representation has served the community well since its inception in the 1970's (Bernstein et al.1) and a large amount of software that uses this representation has been written. However, it is widely recognized that the current PDB format cannot express adequately the large amount of data (content) associated with a single macromolecular structure and the experiment from which it was derived in a way (context) that is consistent and permits direct comparison with other structure entries. Structure comparison, for such purposes as better understanding biological function, assisting in the solution of new structures, drug design, and structure prediction, becomes increasingly valuable as the number of macromolecular structures continues to grow at a near exponential rate. It could be argued that the description of the required content of a structure submission could be met by additional PDB record types. However, this format does not permit the maintenance of the automated level of consistency, accuracy, and reproducibility required for such a large body of data.
A variety of approaches for improved scientific data representation is being explored (IEEE2). The approach described here, which has been developed under the auspices of the International Union of Crystallography (IUCr), is to extend the Crystallographic Information File (CIF) data representation used for describing small molecule structures and associated diffraction experiments. This extension is referred to as the macromolecular Crystallographic Information File (mmCIF) and is the subject of this paper. The paper briefly covers the history of mmCIF, similarities to and differences from the PDB format, contents of the mmCIF dictionary, and how to represent structures using mmCIF. The mmCIF home page (mmCIF3) contains a historic description of the development of the dictionary, current versions of the dictionary in text and HTML formats, software tools, archives of the mmCIF discussion list, and a detailed on-line tutorial (Bourne4).
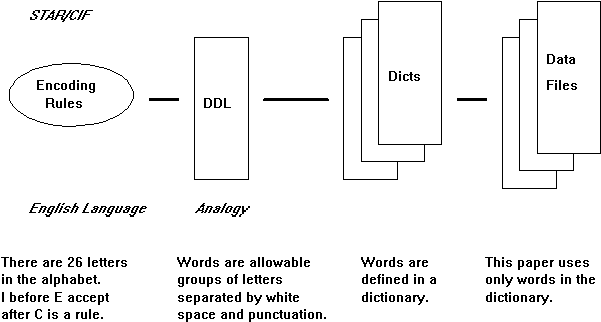
CIF was developed to describe small molecule organic structures and the crystallographic experiment by the International Union of Crystallography (IUCr) Working Party on Crystallographic Information at the behest of the IUCr Commission on Crystallographic Data and the IUCr Commission on Journals. The result of this effort was a core dictionary of data items sufficient for archiving the small molecule crystallographic experiment and its results (Hall et al.5, IUCr6). This core dictionary was adopted by the IUCr at its 1990 Congress in Bordeaux. The format of the small molecule CIF dictionary and the data files based upon that dictionary conform to a restricted version of the Self Defining Text Archive and Retrieval (STAR) representation developed by Hall (Cook and Hall7, Hall and Spadaccini8). STAR permits a data organization that may be understood by analogy with a spoken language (Fig. 1).
Figure 1 Components of the STAR/CIF data representation and their analogy to a natural language.
STAR defines a set of encoding rules similar to saying the English language is comprised of 26 letters. A Dictionary Definition Language (DDL) is defined which uses those rules and which provides a framework from which to define a dictionary of the terms needed by the discipline. Think of the DDL as a computer readable way of declaring that words are made up of arbitrary groups of letters and that words are organized into sentences and paragraphs. The DDL provides a convention for naming and defining data items within the dictionary, declaring specific attributes of those data items, for example, a range of values and the data type, and for declaring relationships between data items. In other words, the DDL defines the format of the dictionary and any new words that are added must conform to that format. Just as words are constantly being added to a language, data items will be added to the dictionaries as the discipline evolves. The STAR encoding rules and the DDL are being used to develop a variety of dictionaries and reference files, for example, the powder diffraction dictionary, the modulated structures dictionary, a file of ideal geometry for amino acids, and an NMR dictionary. This extensibility is attractive since the same basic reading and browsing software (context-based tools) can be used irrespective of the data content. Data files (this paper is an example in our language analogy) are composed of data items found in the dictionaries.
In 1990, the IUCr formed a working group to expand the core dictionary to include data items relevant to the macromolecular crystallographic experiment. Version 1.0 of the mmCIF dictionary (Fitzgerald et al.9, mmCIF3), which encompasses many data items from the current core dictionary (IUCr10), is in the final stage of review by COMCIFs, the IUCr appointed committee overseeing CIF developments. This dictionary has been written using DDL v2.1.1 (Westbrook and Hall11), which is significantly enhanced, yet upwardly compatible with DDL v1.4 (IUCr12) currently used for the small molecule dictionary.
In developing version 1.0 of the mmCIF dictionary we made the following
decisions.
Based on the above, a mmCIF dictionary with approximately 1500 data items (including those data items taken from the small molecule dictionary) was developed. It is not expected that all relevant data items will be present in each mmCIF data file. What data items are mandatory to describe the structure and experiment adequately needs to be decided by community consensus.
The format of a mmCIF containing structural data can best be introduced through analogy with the existing PDB format. A PDB file consists of a series of records each identified by a keyword (e.g., HEADER, COMPND) of up to 6 characters. The format and content of fields within a record are dependent on the keyword. A mmCIF, on the other hand, always consists of a series of name-value pairs (a data item) defined by STAR, where the data name is preceded by a leading underscore (_) to distinguish it from the data value. Thus, every field in a PDB record is represented in mmCIF by a specific data name. The PDB HEADER record,
HEADER PLANT SEED PROTEIN 11-OCT-91 1CBN
becomes:
_struct.entry_id '1CBN'
_struct.title 'PLANT SEED PROTEIN'
_struct_keywords.entry_id '1CBN'
_struct_keywords.text 'plant seed protein'
_database_2.database_id 'PDB'
_database_2.database_code '1CBN'
_database_PDB_rev.rev_num 1
_database_PDB_rev.date_original '1991-10-11'
The name-value pairing represents a major departure from the PDB file format and has the advantage of providing an explicit reference to each item of data within the data file, rather than having the interpretation left to the software reading the file. The name matches an entry in the mmCIF dictionary where characteristics of that data item are explicitly defined. Where multiple values for the same data item exist, the name of the data item or items concerned is declared in a header and the associated values follow in strict rotation. This is a STAR rule referred to as a loop_ construct. This loop_ construct is illustrated in the representation of atomic coordinates.
loop_
_atom_site.group_PDB
_atom_site.type_symbol
_atom_site.label_atom_id
_atom_site.label_comp_id
_atom_site.label_asym_id
_atom_site.label_seq_id
_atom_site.label_alt_id
_atom_site.cartn_x
_atom_site.cartn_y
_atom_site.cartn_z
_atom_site.occupancy
_atom_site.B_iso_or_equiv
_atom_site.footnote_id
_atom_site.auth_seq_id
_atom_site.id
ATOM N N VAL A 11 . 25.369 30.691 11.795 1.00 17.93 . 11 1
ATOM C CA VAL A 11 . 25.970 31.965 12.332 1.00 17.75 . 11 2
ATOM C C VAL A 11 . 25.569 32.010 13.881 1.00 17.83 . 11 3
# [data omitted]
Note that the name construct is of the form _category.extension. The category explicitly defines a natural grouping of data items such that all data items of a single category are contained within a single loop_. There is no restriction on the length of name, beyond the record length limit of 80 characters mentioned below, and while there is no formal syntax within name beyond the category and extension separated by a period, by convention the category and extension are represented as an informal hierarchy of parts, with each part separated by an underscore (_). The components of _atom_site.label are examples.
Questions that arise concerning the separation of data names and data values are solved with some additional syntax. For example, what if the data value contains white space, an underscore, or runs over several lines? Similarly, what if a value in a loop_ is undefined or has no meaning in the context in which it is defined? The following syntax rules, which are a more restricted set of rules than permitted by STAR, complete the mmCIF description.
Comments are preceded by a hash (#) and terminated by a new line.
Data values on a single line may be delimited by pairs of single (') or double (") quotes.
Data values that extend beyond a single line are enclosed within semicolons (;) as the first character of the line that begins the text block and the first character of the line following the last line of text.
Data values which are unknown are represented by a question mark (?).
Data values which are undefined are represented by a period (.).
The length of a record in mmCIF is restricted to 80 characters.
Only printable ASCII characters are permitted.
Only a single level of loop_ is permissible.
To complete the introductory picture of the appearance of a mmCIF data file consider the notion of scope. A PDB file has essentially one form of scope - the complete file. Thus, a single structure or an ensemble of structures is represented by a single file with each member of the ensemble separated by a PDB MODEL keyword record. There is no computer readable mechanism for associating components of say the REMARK records with a particular member of the ensemble. The mmCIF representation deals with this issue by using the STAR data block concept. Data blocks begin with data_ and have a scope that extends until the next data_ or an end-of-file is reached. A name may appear only once in a data block, but data items may appear in any order. A consequence of these STAR rules is that the combination of data block name and data name is always unique.
Table I summarizes the category groups, their associated individual categories and their definitions as found in the mmCIF dictionary version 0.8.02 dated March 18, 1996. This comprehensive hierarchy of categories follows closely the progress of the experiment and the subsequent structure description.
The categories describing the crystallographic experiment are relatively self explanatory and will not be detailed here. We will, however, outline the data model used to describe the resulting structure and its description.
The structural data model can most simply be described as containing three interrelated groups of categories: ATOM_SITE categories that give coordinates and related information of the structure; ENTITY categories, which describe the chemistry of the components of the structure, and STRUCT categories, which analyze and describe the structure.
The data items in the ATOM_SITE category record details about the atom sites including the coordinates, the thermal displacement parameters, the errors in the parameters and include a specification of the component of the asymmetric unit to which an atom belongs.
The ENTITY category categorizes the unique chemical components of the asymmetric unit as to whether they are polymer, non-polymer or water. The characteristics of a polymer are described by the ENTITY_POLY category and the sequence of the chemical components comprising the polymer by the ENTITY_POLY_SEQ category. The CHEM_COMP categories describe the standard geometries of the monomer units such as the amino acids and nucleotides as well as that of the ligands and solvent groups.
The STRUCT_BIOL category allows the person to describe
the biologically relevant features of a structure and its component parts.
The STRUCT_BIOL_GEN category provides the information about how
to generate the biological unit from the components of the asymmetric unit
which are in turn specified by the STRUCT_ASYM category. Various
features of the structure such as intermolecular hydrogen bonds, special
sites and secondary structure are specified in STRUCT_CONN, STRUCT_SITE
and STRUCT_CONF, respectively. Figure 2 illustrates the interrelationships
among these categories.
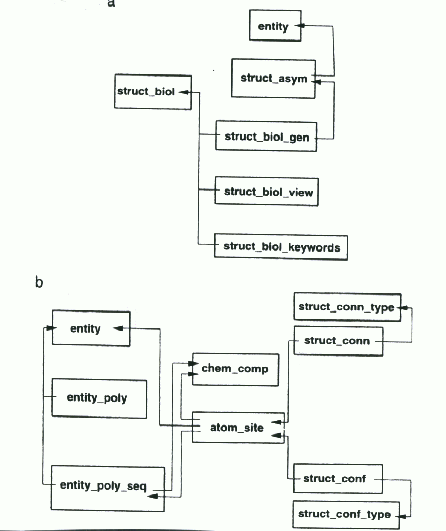
Figure 2 a) The relationships between categories which describe
biologically relevant structure. b) The relationships between categories
describing polymer structure, the atomic coordinates, and those categories
which describe structural features such as hydrogen bonding and secondary
structure.
These and other major descriptive features of the mmCIF dictionary are best explored by example. A browsable dictionary can be found at the mmCIF WWW site (mmCIF3) as well as some complete examples. Complete examples for all nucleic acids can be found at the Nucleic Acid Database WWW site (NDB13). Partial mmCIFs for every structure in the PDB are available at two WWW sites (PDB14, SDSC15) having been generated with the program pdb2cif (Bernstein et al.16).
Example One
Starting simply, consider the protein crambin which is a single polypeptide chain of 48 residues and in the low temperature form at 0.83 Å resolution (Teeter et al.17; PDB code 1CBN) has nearly all the protein bound solvent resolved as well as an ethanol molecule co-crystallized. The protein shows recognizable sequence micro heterogeneity at positions 22 (Pro/Ser) and 25 (Leu/Ile) and 24% of residues show discrete disorder. While these features are described using data items in the mmCIF dictionary, they are not detailed here for the sake of simplicity.
Since the biological function of this molecule is unknown, no biologically relevant structural components are justified. A single identifier (crambin_1) is used to identify the unknown biological function of this molecule.
_struct_biol.id crambin_1
_struct_biol.details
; The function of this protein is unknown and therefore the
biological unit is assumed to be the single polypeptide
chain without co-crystallization factors i.e. ethanol.
;
The single biological descriptor, crambin_1, is generated from the single polypeptide chain found in the asymmetric unit without any symmetry transformations applied. The polypeptide chain is designated chain_a.
_struct_biol_gen.biol_id crambin_1
_struct_biol_gen.asym.id chain_a
_struct_biol_gen.symmetry 1_555
The chemical components of the asymmetric unit are three entities: a single polypeptide chain characterized as a polymer, ethanol characterized as non-polymer, and water. Whether the source of the entity is a natural product, or it has been synthesized is also indicated.
loop_
_entity.id
_entity.type
_entity.formula_weight
_entity.src_method
A polymer 4716 'NATURAL'
ethanol non-polymer 52 'SYNTHETIC'
H20 water 18 .
It is then possible to expand upon this basic description of each entity using the entity.id as a reference. So for example the common and systematic names are specified as,
_entity_name_com.entity_id A
_entity_name_com.name crambin
_entity_name_sys.entity_id A
_entity_name_sys.name 'Crambe Abyssinica'
Similarly, the natural and synthetic description can be given in more detail, so for the natural product we have,
_entity_src_nat.entity_id A
_entity_src_nat.common_name 'Abyssinian Cabbage'
_entity_src_nat.genus ?
_entity_src_nat.species ?
_entity_src_nat.details ?
Using the entities as building blocks the contents of the asymmetric unit are specified. Crambin is straightforward since each entity appears only once in the asymmetric unit.
loop_
_struct_asym.id
_struct_asym.entity_id
_struct_asym.details
chain_a A 'Single polypeptide chain'
ethanol ethanol 'Cocrystallized ethanol molecule'
H20 H20 .
Entities classified as polymer, in this instance only that entity identified as A, is further described. First, the overall features of the polypeptide chain.
_entity_poly.entity_id A
_entity_poly.type polypeptide(L)
_entity_poly.nstd_chirality no
_entity_poly.nstd_linkage no
_entity_poly.nstd_monomers no
_entity_poly.type_details 'Microheterogeneity at 22 and 25'
and then the component parts,
loop_
_entity_poly_seq.entity_id
_entity_poly_seq.num
_entity_poly_seq.mon_id
A 1 THR A 2 THR
# [data omitted]
A 22 PRO A 23 GLU
A 24 ALA A 25 LEU
# [data omitted]
A 47 ALA A 48 ASN
The entity may also exist in other databases and these references may be cited and described. For the entity designated A, which is defined in Genbank but without sequence microheterogeneity we have,
loop_
_struct_ref.id
_struct_ref.entity_id
_struct_ref.biol_id
_struct_ref.db_name
_struct_ref.db_code
_struct_ref.seq_align
_struct_ref.seq_dif
_struct_ref.details
1 A crambin_1 'Genbank' '493916' 'entire' 'no' .
2 A crambin_1 'PDB' '1CBN' 'entire' 'no' .
Once each polymer entity is defined, the details of the secondary structure are defined using the STRUCT_CONF category.
loop_
_struct_conf.id
_struct_conf.conf_type.id
_struct_conf.beg_label_comp_id
_struct_conf.beg_label_asym_id
_struct_conf.beg_label_seq_id
_struct_conf.end_label_comp_id
_struct_conf.end_label_asym_id
_struct_conf.end_label_seq_id
_struct_conf.details
H1 HELX_RH_AL_P ILE chain_a 7 PRO chain_a 19 'HELX-RH3T 17-19'
H2 HELX_RH_AL_P GLU chain_a 23 THR chain_a 30 'Alpha-N start'
S1 STRN_P CYS chain_a 32 ILE chain_a 35 .
S2 STRN_P THR chain_a 1 CYS chain_a 4 .
S3 STRN_P ASN chain_a 46 ASN chain_a 46 .
S4 STRN_P THR chain_a 39 PRO chain_a 41 .
T1 TURN-TY1_P ARG chain_a 17 GLY chain_a 20 .
T2 TURN-TY1_P PRO chain_a 41 TYR chain_a 44 .
These assignments are further enumerated over those made in a PDB file for the record types HELIX, TURN and SHEET. Moreover, the STRUCT_CONF_TYPE category (Table I) specifies the method of assignment which could, for example, be deduced by the crystallographer from the electron density maps or defined algorithmically.
loop_
_struct_conf_type.id
_struct_conf_type.criteria
_struct_conf_type.reference
HELX_RH_AL_P 'author judgement' .
STRN_P 'author judgement' .
TURN_TY1_P 'author judgement' .
# HELX_RH_P 'Kabsch and Sander' 'Biopolymers (1983) 22:2577'
The commented entry at the end is a hypothetical example for a calculated assignment. Data items also exist (Table I) for the description of beta sheets, but are not shown in this introductory example.
Interactions between various portions of the structure are described by the STRUCT_CONN and associated STRUCT_CONN_TYPE category.
loop_
struct_conn.id
struct_conn.conn_type_id
struct_conn.ptnr1_label_comp_id
struct_conn.ptnr1_label_asym_id
struct_conn.ptnr1_label_seq_id
struct_conn.ptnr1_label_atom_id
struct_conn.ptnr1_role
struct_conn.ptnr1_symmetry
struct_conn.ptnr2_label_comp_id
struct_conn.ptnr2_label_asym_id
struct_conn.ptnr2_label_seq_id
struct_conn.ptnr2_label_atom_id
struct_conn.ptnr2_role
struct_conn.ptnr2_symmetry
struct_conn.details
SS1 disulf CYS chain_a 3 S 1_555 CYS chain_a 40 S 1_555 .
SS2 disulf CYS chain_a 4 S 1_555 CYS chain_a 32 S 1_555 .
# [data omitted]
HB1 hydrog SER chain_a 6 OG positive 1_555 .
LEU chain_a 8 O negative 1_556 .
HB2 hydrog ARG chain_a 17 N positive 1_555 .
ASP chain_a 43 O negative 1_554 .
# [data omitted]
These intermolecular interactions are partially specified on PDB CONNECT records. However mmCIF provides an additional level of detail such that the criteria used to define an interaction may be given using the STRUCT_CONN_TYPE category. Here is a hypothetical example used to describe a salt bridge and a hydrogen bond.
loop_
_struct_conn_type.id
_struct_conn_type.criteria
_struct_conn_type.reference
saltbr 'negative to positive distance > 2.5 \%A and < 3.2 \%A ' .
hydrog 'N to O distance > 2.5 \%A, < 3.2 \%A, NOC angle < 120°' .
Example Two
Consider a mmCIF representation for a more complex structure. The gene regulatory protein 434 CRO complexed with a 20 base pair DNA segment containing operator (Mondragon and Harrison18; PDB code 3CRO).
loop_
_struct_biol.id
_struct_biol.details
complex
; The complex consists of 2 protein domains bound to a
20 base pair DNA segment.
;
protein
; Each of the 2 protein domains is a single homologous
polypeptide chain of 71 residues designated L and R.
;
DNA
; The two strands (A and B) are complementary given a one
base offset.
;
The protein/DNA complex, the protein, and the DNA are considered as three separate biological components each generated from the contents of the asymmetric unit. No crystallographic symmetry need be applied to generate the biologically relevant components.
loop_
_struct_biol_gen.biol_id
_struct_biol_gen.asym.id
_struct_biol_gen.symmetry
complex L 1_555
complex R 1_555
complex A 1_555
complex B 1_555
protein L 1_555
protein R 1_555
DNA A 1_555
DNA B 1_555
loop_
_entity.id
_entity.type
dimer polymer
DNA_A polymer
DNA_B polymer
water water
Since each protein domain is chemically identical they constitute a single entity which has been designated dimer. The complementary DNA strands are not chemically identical and therefore constitute two separate entities:
_struct_asym.id
_struct_asym.entity_id
_struct_asym.details
L dimer '71 residue polypeptide chain'
R dimer '71 residue polypeptide chain'
A DNA_A '20 base strand'
B DNA_B '20 base strand'
H2O water 'solvent'
Features of the CRO 434 secondary structure and intermolecular contacts can be described in the same way in which crambin was represented and are not repeated.
In preparing these examples of representing macromolecular structure using mmCIF it was necessary to return to the original papers since not all the relevant information could be retrieved from the PDB entry. This is evidence that mmCIF provides additional information which also has the advantage of being in a computer readable form. The consequence is that it places additional emphasis on the person preparing the mmCIF. It is anticipated that full use of the expressive power of mmCIF will only be made when existing structure solution and refinement programs are modified to maintain mmCIF data items and software tools exist to help prepare and use a mmCIF effectively. A variety of software tools have been developed for mmCIF (Bernstein, et al. 16; Westbrook, et al. 19). A description of a variety of other efforts can be found elsewhere (Bourne20). Code and documentation is available at the mmCIF WWW site (mmCIF3). A long term goal might be to maintain all aspects of the structure determination in an electronic laboratory notebook that uses mmCIF as its underlying data representation. The notebook would have a "journal" button that would be used at the appropriate time.
The development of the mmCIF dictionary has been a community effort.
1. F.C. Bernstein, T.F. Koetzle, G.J.B. Williams, E.F. Meyer,Jr., M.D. Brice, J.R.Rogers, O. Kennard, T. Shimanouchi, and M. Tasumi, J. Mol. Biol. 112, 535 (1977).
2. IEEE Metadata. http://www.llnl.gov/liv_comp/metadata/ (1996).
3. mmCIF. http://ndbserver.rutgers.edu/NDB/mmcif/ (1996).
4. P.E. Bourne. http://www.sdsc.edu/pb/cif/overview.html (1996).
5. S.R. Hall, F.H. Allen, and I.D. Brown, Acta Cryst. A47, 655 (1991).
6. IUCr. ftp://ftp.iucr.ac.uk/cifdics/cifdic.c91 US mirror (1996).
7. A. Cook and S.R. Hall, J. Chem Inf. Comput. Sci. 31, 326 (1992).
8. S.R. Hall and N.Spadaccini, J. Chem. Inf. Comput. Sci. 34, 505 (1994).
9. P.M.D. Fitzgerald, H.M. Berman, P.E. Bourne, B. McMahon, K. Watenpaugh, and J.D. Westbrook Acta Cryst. A52 Sup., C575 (1996).
10. IUCr. ftp://ftp.iucr.ac.uk/cifdics/cifdic.c96 US mirror (1996).
11. J.D. Westbrook and S.R. Hall. http://ndbserver.rutgers.edu/mmcif/ddl/ (1995).
12. IUCr. ftp://ftp.iucr.ac.uk/cifdics/ddldic.c95 US mirror (1995).
13. NDB. http://ndbserver.rutgers.edu/ (1996).
14. PDB. http://www.pdb.bnl.gov/cgi-bin/pdbmain (1996).
15. SDSC. http://www.sdsc.edu/moose (1996).
16. H.J. Bernstein, F.C. Bernstein, and P.E. Bourne. In preparation (1996).
17. M.M.Teeter, S.M. Roe, and N. Ho Heo, J. Mol. Biol. 230, 292 (1993).
18. A.Mondragon and S.C.Harrison, J Mol. Biol. 219, 321 (1991).
19. J.D. Westbrook, S.H. Hsieh, and P.M.D. Fitzgerald, J. App. Cryst. In press (1996).
20. P.E.Bourne (Ed.), Proceedings of the first
macromolecular CIF tools workshop. Tarrytown NY (1993).
Table 1 The mmCIF category groups and associated categories taken from the mmCIF dictionary
| CATEGORY GROUPS AND MEMBERS | DEFINITION |
| INCLUSIVE GROUP | All category groups |
| ATOM GROUP | |
| ATOM_SITE | Details of each atomic position |
| ATOM_SITE_ANISOTROP | Anisotropic thermal displacement |
| ATOM_SITES | Details pertaining to all atom sites |
| ATOM_SITES_ALT | Details pertaining to alternative atoms sites as found in disorder etc. |
| ATOM_SITES_ALT_ENS | Details pertaining to alternative atoms sites as found in ensembles e.g. from NMR and modeling experiments |
| ATOM_SITES_ALT_GEN | Generation of ensembles from multiple conformations |
| ATOM_SITES_FOOTNOTE | Comments concerning one or more atom sites |
| ATOM_TYPE | Properties of an atom at a particular atom site |
| AUDIT GROUP | |
| AUDIT | Detail on the creation and updating of the mmCIF |
| AUDIT_AUTHOR | Author(s) of the mmCIF including address information |
| AUDIT_CONTACT_AUTHOR | Author(s) to be contacted |
| CELL GROUP | |
| CELL | Unit cell parameters |
| CELL_MEASUREMENT | How the cell parameters were measured |
| CELL_MEASUREMENT_REFLN | Details of the reflections used to determine the unit cell parameters |
| CHEM_COMP GROUP | |
| CHEM_COMP | Details of the chemical components |
| CHEM_COMP_ANGLE | Bond angles in a chemical component |
| CHEM_COMP_ATOM | Atoms defining a chemical component |
| CHEM_COMP_BOND | Characteristics of bonds in a chemical component |
| CHEM_COMP_CHIR | Details of the chiral centers in a chemical component |
| CHEM_COMP_CHIR_ATOM | Atoms comprising a chiral center in a chemical component |
| CHEM_COMP_LINK | Linkages between chemical groups |
| CHEM_COMP_PLANE | Planes found in a chemical component |
| CHEM_COMP_PLANE_ATOM | Atoms comprising a plane in a chemical component |
| CHEM_COMP_TOR | Details of the torsion angles in a chemical component |
| CHEM_COMP_TOR_VALUE | Target values for the torsion angles in a chemical component |
| CHEM_LINK GROUP | |
| CHEM_LINK | Details of the linkages between chemical components |
| CHEM_LINK_ANGLE | Details of the angles in the chemical component linkage |
| CHEM_LINK_BOND | Details of the bonds in the chemical component linkage |
| CHEM_LINK_CHIR | Chiral centers in a link between two chemical components |
| CHEM_LINK_CHIR_ATOM | Atoms bonded to a chiral atom in a linkage between two chemical components |
| CHEM_LINK_PLANE | Planes in a linkage between two chemical components |
| CHEM_LINK_PLANE_ATOM | Atoms in the plane forming a linkage between two chemical components |
| CHEM_LINK_TOR | Torsion angles in a linkage between two chemical components |
| CHEM_LINK_TOR_VALUE | Target values for torsion angles enumerated in a linkage between two chemical components |
| CHEMICAL GROUP | |
| CHEMICAL | Composition and chemical properties |
| CHEMICAL_CONN_ATOM | Atom position for 2-D chemical diagrams |
| CHEMICAL_CONN_BOND | Bond specifications for 2-D chemical diagrams |
| CHEMICAL_FORMULA | Chemical formula |
| CITATION GROUP | |
| CITATION | Literature cited in reference to the data block |
| CITATION_AUTHOR | Author(s) of the citations |
| CITATION_EDITOR | Editor(s) of citations where applicable |
| COMPUTING GROUP | |
| COMPUTING | Computer programs used in the structure analysis |
| SOFTWARE | More detailed description of the software used in the structure analysis |
| DATABASE GROUP | |
| DATABASE | Superseded by DATABASE_2 |
| DATABASE_2 | Codes assigned to mmCIFs by maintainers of recognized databases |
| DATABASE_PDB_CAVEAT | CAVEAT records originally found in the PDB version of the mmCIF data file |
| DATABASE_PDB_MATRIX | MATRIX records originally found in the PDB version of the mmCIF data file |
| DATABASE_PDB_REMARK | REMARK records originally found in the PDB version of the mmCIF data file |
| DATABASE_PDB_REV | Taken from the PDB REVDAT records |
| DATABASE_PDB_REV_RECORD | Taken from the PDB REVDAT records |
| DATABASE_PDB_TVECT | TVECT records originally found in the PDB version of the mmCIF data file |
| DIFFRN GROUP | |
| DIFFRN | Details of diffraction data and the diffraction experiment |
| DIFFRN_ATTENUATOR | Diffraction attenuator scales |
| DIFFRN_MEASUREMENT | Details on how the diffraction data were measured |
| DIFFRN_ORIENT_MATRIX | Orientation matrices used when measuring data |
| DIFFRN_ORIENT_REFLN | Reflections that define the orientation matrix |
| DIFFRN_RADIATION | Details on the radiation and detector used to collect data |
| DIFFRN_REFLN | Unprocessed reflection data |
| DIFFRN_REFLNS | Details pertaining to all reflection data |
| DIFFRN_SCALE_GROUP | Details of reflections used in scaling |
| DIFFRN_STANDARD_REFLN | Details of the standard reflections used during data collection |
| DIFFRN_STANDARDS | Details pertaining to all standard reflections |
| ENTITY GROUP | |
| ENTITY | Details pertaining to each unique chemical component of the structure |
| ENTITY_KEYWORDS | Keywords describing each entity |
| ENTITY_LINK | Details of the links between entities |
| ENTITY_NAME_COM | Common name for the entity |
| ENTITY_NAME_SYS | Systematic name for the entity |
| ENTITY_POLY | Characteristics of a polymer |
| ENTITY_POLY_SEQ | Sequence of monomers in a polymer |
| ENTITY_SRC_GEN | Source of the entity |
| ENTITY_SRC_NAT | Details of the natural source of the entity |
| ENTRY GROUP | |
| ENTRY | Identifier for the data block |
| EXPTL GROUP | |
| EXPTL | Experimental details relating to the physical properties of the material, particularly absorption |
| EXPTL_CRYSTAL | Physical properties of the crystal |
| EXPTL_CRYSTAL_FACE | Details pertaining to the crystal faces |
| EXPTL_CRYSTAL_GROW | Conditions and methods used to grow the crystals |
| EXPTL_CRYSTAL_GROW_COMP | Components of the solution from which the crystals were grown |
| GEOM GROUP | |
| GEOM | Derived geometry information |
| GEOM_ANGLE | Derived bond angles |
| GEOM_BOND | Derived bonds |
| GEOM_CONTACT | Derived intermolecular contacts |
| GEOM_TORSION | Derived torsion angles |
| JOURNAL GROUP | |
| JOURNAL | Used by journals and not the mmCIF preparer |
| PHASING GROUP | |
| PHASING | General phasing information |
| PHASING_AVERAGING | Phase averaging of multiple observations |
| PHASING_ISOMORPHOUS | Phasing information from an isomorphous model |
| PHASING_MAD | Phasing via multiwavelength anomolous dispersion (MAD) |
| PHASING_MAD_CLUST | Details of a cluster of MAD experiments |
| PHASING_MAD_EXPT | Overall features of the MAD experiment |
| PHASING_MAD_RATIO | Ratios between pairs of MAD datasets |
| PHASING_MAD_SET | Details of individual MAD datasets |
| PHASING_MIR | Phasing via single and multiple isomorphous replacement |
| PHASING_MIR_DER | Details of individual derivatives used in MIR |
| PHASING_MIR_DER_REFLN | Details of calculated structure factors |
| PHASING_MIR_DER_SHELL | As above but for shells of resolution |
| PHASING_MIR_DER_SITE | Details of heavy atom sites |
| PHASING_MIR_SHELL | Details of each shell used in MIR |
| PHASING_SET | Details of data sets used in phasing |
| PHASING_SET_REFLN | Values of structure factors used in phasing |
| PUBL GROUP | |
| PUBL | Used when submitting a publication as a mmCIF |
| PUBL_AUTHOR | Authors of the publication |
| PUBL_MANUSCRIPT_INCL | To include special data names in the processing of the manuscript |
| REFINE GROUP | |
| REFINE | Details of the structure refinement |
| REFINE_B_ISO | Details pertaining to the refinement of isotropic B values |
| REFINE_HIST | History of the refinement |
| REFINE_LS_RESTR | Details pertaining to the least squares restraints used in refinement |
| REFINE_LS_SHELL | Results of refinement broken down by resolution |
| REFINE_OCCUPANCY | Details pertaining to the refinement of occupancy factors |
| REFLN GROUP | |
| REFLN | Details pertaining to the reflections used to derive the atom sites |
| REFLNS | Details pertaining to all reflections |
| REFLNS_SCALE | Details pertaining to scaling factors used with respect to the structure factors |
| REFLNS_SHELL | As REFLNS, but by shells of resolution |
| STRUCT GROUP | |
| STRUCT | Details pertaining to a description of the structure |
| STRUCT_ASYM | Details pertaining to structure components within the asymmetric unit |
| STRUCT_BIOL | Details pertaining to components of the structure that have biological significance |
| STRUCT_BIOL_GEN | Details pertaining to generating biological components |
| STRUCT_BIOL_KEYWORDS | Keywords for describing biological components |
| STRUCT_BIOL_VIEW | Description of views of the structure with biological significance |
| STRUCT_CONF | Conformations of the backbone |
| STRUCT_CONF_TYPE | Details of each backbone conformation |
| STRUCT_CONN | Details pertaining to intermolecular contacts |
| STRUCT_CONN_TYPE | Details of each type of intermolecular contact |
| STRUCT_KEYWORDS | Description of the chemical structure |
| STRUCT_MON_DETAILS | Calculation summaries at the monomer level |
| STRUCT_MON_NUCL | Calculation summaries specific to nucleic acid monomers |
| STRUCT_MON_PROT | Calculation summaries specific to protein monomers |
| STRUCT_MON_PROT_CIS | Calculation summaries specific to cis peptides |
| STRUCT_NCS_DOM | Details of domains within an ensemble of domains |
| STRUCT_NCS_DOM_LIM | Beginning and end points within polypeptide chains forming a specific domain |
| STRUCT_NCS_ENS | Description of ensembles |
| STRUCT_NCS_ENS_GEN | Description of domains related by non-crystallographic symmetry |
| STRUCT_NCS_OPER | Operations required to superimpose individual members of an ensemble |
| STRUCT_REF | External database references to biological units within the structure |
| STRUCT_REF_SEQ | Describes the alignment of the external database sequence with that found in the structure |
| STRUCT_REF_SEQ_DIF | Describes differences in the external database sequence with that found in the structure |
| STRUCT_SHEET | Beta sheet description |
| STRUCT_SHEET_HBOND | Hydrogen bond description in beta sheets |
| STRUCT_SHEET_ORDER | Order of residue ranges in beta sheets |
| STRUCT_SHEET_RANGE | Residue ranges in beta sheets |
| STRUCT_SHEET_TOPOLOGY | Topology of residue ranges in beta sheets |
| STRUCT_SITE | Details pertaining to specific sites within the structure |
| STRUCT_SITE_GEN | Details pertaining to how the site is generated |
| STRUCT_SITE_KEYWORDS | Keywords describing the site |
| STRUCT_SITE_VIEW | Description of views of the specified site |
| SYMMETRY GROUP | |
| SYMMETRY | Details pertaining to space group symmetry |
| SYMMETRY_EQUIV | Equivalent positions for the specified space group |
[an error occurred while processing this directive]